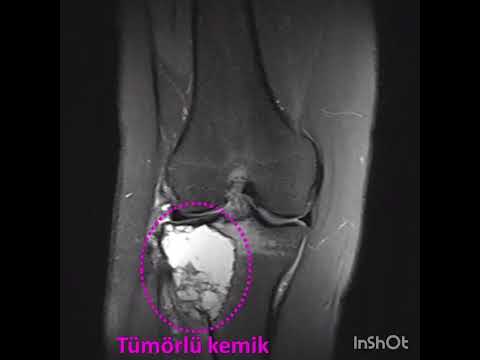
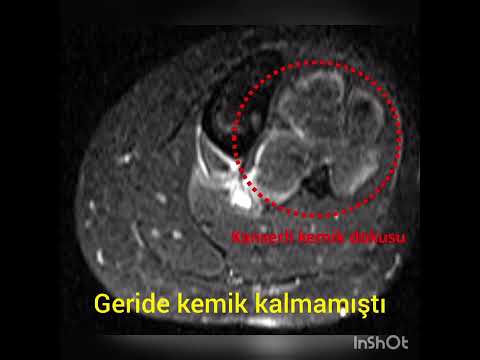
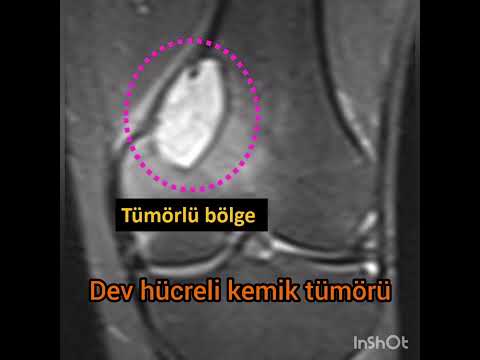
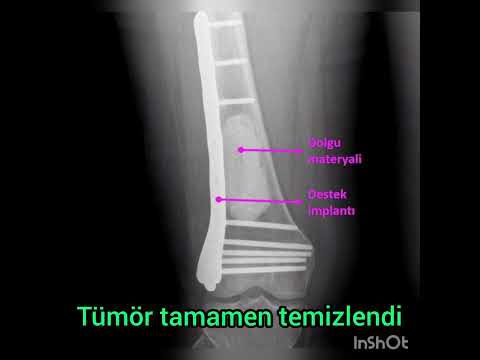
لغة
لغة Türkçe
Türkçe English
English Arabic
Arabic Germany
Germany Russian
RussianSinir ve Damar Kökenli Yumuşak Doku Sarkomları

ÖZET
Kötü
huylu sinir kılıfı tümörleri; Kötü huylu periferik sinir kılıfı tümörü (MPSKT),
Kötü huylu granüler hücreli tümör ve Ektomezenkimoma olarak
sınıflandırılmaktadır. MPSKT dışındaki tü[1]mörler oldukça ender
görülmektedir. MPSKT, tüm yumuşak doku sarkomları içerisinde %5 gibi küçük bir
orana sahiptir. Tipik olarak 20-50 yaşları arasında görülür ve kadınlar
erkeklere oranla daha fazla etkilenmektedir. Alt ekstremiteler lezyonların en
sık rastlandığı yerlerdir. Çoğu MPSKT siyatik sinir gibi büyük periferal sinirleri
tutar. Hastalarda gittikçe büyüyen kitle varlığıyla beraber nöropatik
semptomlar da gözlenebilir. Görüntülemede manyetik rezonans (MR) öncelikli
tercihtir. Kötü huylu damar tümörleri ise damar endotelinden kaynaklanan
lezyonlardır. Tüm yaşlarda görül[1]mesine
rağmen yedinci dekatta daha sık rastlanırlar. Çoğu olguda etyoloji belirsizdir
ancak bazı va[1]kaların
radyasyonla ilişkili olduğu bilinmektedir. Sıklıkla alt ekstremitelere
lokalizedir. Dünya Sağlık Örgütü (WHO) kötü huylu damar tümörlerini farklı alt
tiplere ayırmış olmakla beraber ön sırada “anjiyosarkom” gelmektedir. Bu
tümörler oldukça agresif olup, yüksek mortalite oranına sahiptir.
Anahtar Kelimeler: Sinir kılıfı
tümörleri; nörofibrom; vasküler tümörler; hemanjiyosarkom
ABSTRACT
Malignant
neural sheath tumors are categorized as malignant peripheral nerve sheath
tumors (MPNST), malignant granular cell tumors and ectomesenchymoma. Tumors
other than MP[1]NSTs
are rarely seen. MPNSTs comprise a small portion of all soft tissue sarcomas,
which accounts to approximately 5%. They are typically seen between the ages
20-50 and women are affected more frequently than men. Lower extremities are
the most common sites. Most MPNSTs occur in con[1]juction with large
peripheral nerves such as sciatic nerve. Neuropathic symptoms can be also ob[1]served
in patients with growing lesions. Magnetic resonance imaging (MRI) is the
preferred imaging modality. Malignant vascular tumors originate from vascular
endothelium. Peak age of incidence appears to be seventh decade, although they
might be seen in all ages. Generally the etiology is un[1]clear, but it is known that
some cases are associated with radiation. The lesions are often localized to
the lower extremities. The World Health Organization (WHO) has categorized
malignant vas[1]cular
tumors into different subtypes, with angiosarcoma ranking as the most
prominent. These tu[1]mors
are very aggressive and have a high mortality rate
Keywords: Nerve sheath
neoplasms; neurofibroma; vascular neoplasms; hemangiosarcoma
SİNİR KÖKENLİ YUMUŞAK
DOKU SARKOMLARI
Kötü
huylu sinir kılıfı tümörleri; kötü huylu periferik sinir kılıfı tümörü (MPSKT),
kötü huylu granüler hücreli tümör ve ektomezenkimoma olarak
sınıflandırılmaktadır. MPSKT dışındaki tümörler oldukça ender görülmek[1]tedirler.
Ektomezenkimoma, nöronal ya da nöral elemanlar içeren rabdomiyosar[1]komdan
oluşur. Kötü huylu granüler hücreli tümör ise; Schwann hücre fenotipli granüler
hücrelerden oluşmaktadır. MPSKT periferik sinirlerin Schwann hücrele[1]rinden,
perinöral hücrelerden ya da fibroblastik iğsi hücrelerinden köken almakta
Turkiye Klinikleri J Orthop & Traumatol-Special Topics 2017;10(4):325-30
325 dır. Bazı otoriteler tarafından “Malign Schwannoma”, “Nörofibrosarkom” ya
da “Nörojenik Sarkom” gibi isimlerle de anılmıştır. Dünya Sağlık Örgütü (WHO)
bu tümörü sinir gövdesi ya da nörofibromdan kaynaklanan fuziform hücreli sarkom
olarak tanımlar. 1 MPSKT, tüm yumuşak doku sarkomları içerisinde %5 gibi küçük
bir orana sahip ve oldukça kötü huylu bir tümördür. 2 Tipik olarak 20-50 yaş aralığında
görülür. Kadınlar erkeklere oranla daha fazla etkilenmektedir. MPSKT
olgularının yarısı sporadik olarak görülürken, diğer yarısı ailesel geçiş
gösteren, otozomal dominant bir hastalık olan ve 3000 canlı doğumda 1 görülen
nörofib[1]romatozis
tip 1 (NF1) hastalığı ile ilişkilidir. 3,4 Bu olgu[1]larda MPSKT gelişimi
ortalama 1 dekad önce ortaya çıkar. Sporadik hastaların %10’u daha önce
uygulanmış radyoterapi ile ilişkilendirilir. Bu tip bir hastada MPSKT
gelişebilmesi için genellikle 10-20 yıl geçmesi gerekir. Diğerlerinde ise neden
bilinmemektedir. 5 KLİNİK VE GÖRÜNTÜLEME Hastaların kliniğinde genellikle
ağrısız ve gittikçe büyüyen bir kitle söz konusuyken, zamanla bir takım
nörolojik bulgular da olaya eşlik edebilir. Bu süreç uzun bir dönemi
kapsayabilir. NF1’li hastalarda bilinen ve sebat etmiş bir lezyon aniden
büyümeye başlarsa malign transformasyon mutlaka göz önüne alınmalı ve biyopsi
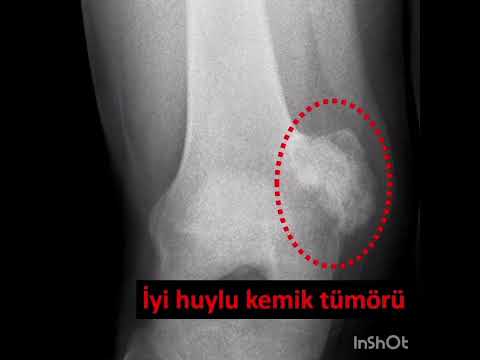
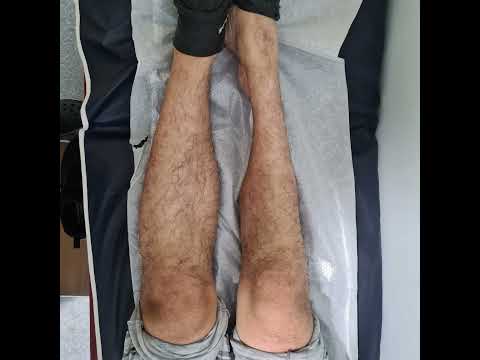
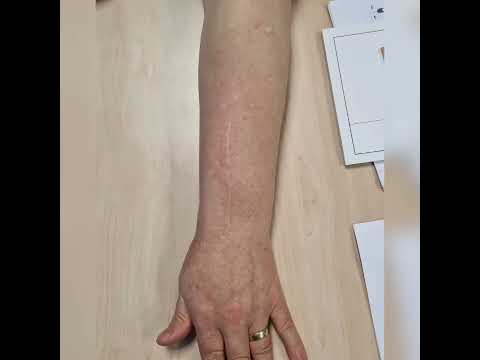
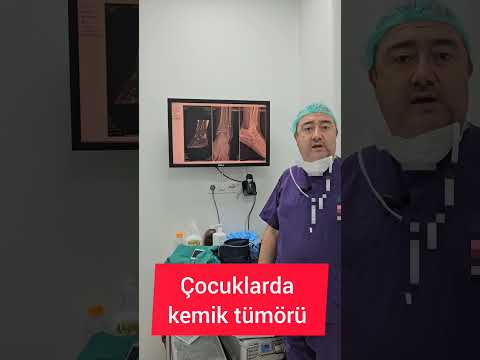
yapılmalıdır. Lezyon, en fazla alt ekstremite ve özellikle siyatik sinirde
görülmektedir (Resim 1). 6 Hasta daha çok oturur pozisyondayken lezyonu fark
eder. Bazı hastalarda parestezi, motor zafiyet ve radikülopati gibi nörolojik
semptomlar da görülebilmekte ve tinel testi pozitif olabilmektedir. Bu bulgular
MPSKT’de benign lezyonlara kıyasla daha yüksek oranda gözlenir. Lezyonlar daha
nadir olarak gövdede ve boyunda da görülebilir. Siyatik sinir ile beraber
sakral pleksus ve brakial pleksus gibi büyük sinirler daha fazla tutulur.
Kutanöz yerleşim ise oldukça nadirdir ve NF1 ile birlikteliği sık değildir.

Kitlenin
görüntülenmesinde standart yumuşak doku kitlelerinde olduğu gibi manyetik
rezonans (MR) tercih edilir. Ancak birçok yumuşak doku sarkomunda olduğu gibi
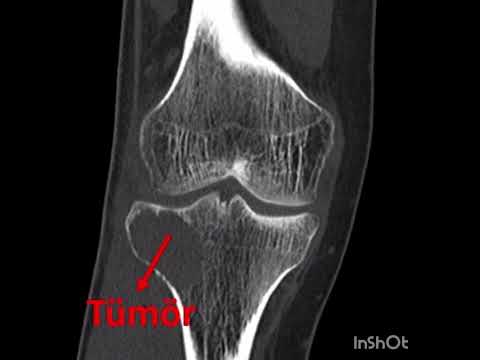
MR’da spesifik bir bulgu görülmemektedir. 5 MR ile tümörün yerleşimi, boyutu,
çevre anatomik ya[1]pılarla
olan ilişkisi ve heterojenitesi rahatça anlaşılabile[1]ceği gibi uygun kesitlerde
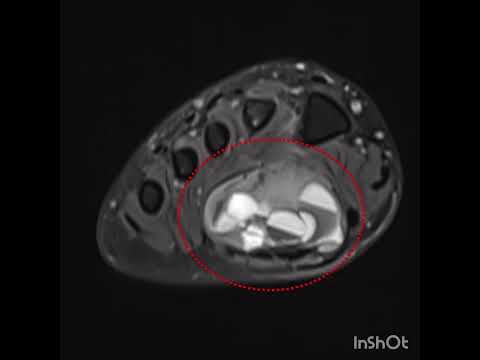
sinirle olan ilişkisi de açıkça görülebilir (Resim 2). NF1’li hastaların
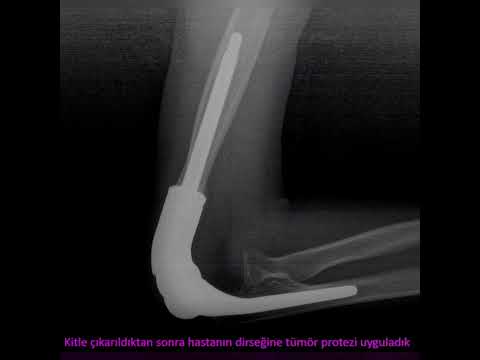
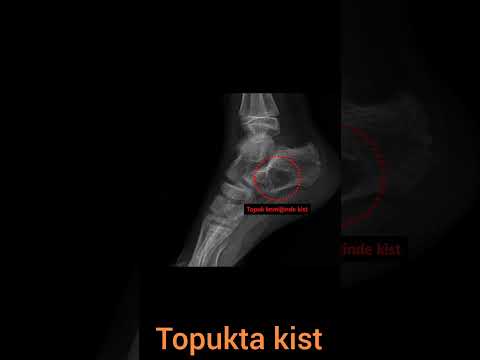
tanısında ise FDG-PET oldukça hassas bir yöntemdir (Resim 3). 8 Kitle çoğu
zaman 5 cm’den büyük ve derin yerleşimlidir. Kesin tanı biyopsi ile konulur ve
tedavi için hastaya geniş rezeksiyon uygulanır. Bu aşamada radyoterapi ve
kemoterapi gibi tedavilerden de yararlanılabilir. Hasta[1]lığın seyrinde çoğu zaman
kötü bir tabloyla karşılaşıl[1]maktadır.
PATOLOJİ VE GENETİK
MPSKT’ler
genellikle büyük çaplı, düzensiz sınır göste[1]ren, solid kitle oluşturan
ve kesit yüzü fokal nekroz, ka[1]nama
veya miksoid değişiklik gösterebilen tümörlerdir. 9 Tipik vakalar mikroskopik
olarak fasiküler gelişim gös[1]teren
iğsi hücrelerden oluşur ve fibrosarkomu andırır. 1,10 Hiperselüler ve
hiposelüler alanlar izlenir. Tümör mito[1]tik
olarak aktiftir. Coğrafik nekroz alanları görülebilir. Tümör hücreleri damarlar
çevresinde yoğunlaşma eğili[1]mindedir.
Epitelioid hücre morfolojisi gösterebilir. Yük[1]sek dereceli andiferansiye
pleomorfik sarkomu andıraca

şekilde
yaygın pleomorfizm içerebilir. İskelet kası, kemik, kıkırdak gibi heterolog
elemanlar bulunabilir. İs[1]kelet
kası diferansiyasyonu gösteren MPSKT’ye ‘Malign triton tümör’ denir. 1,9 MPSKT
olgularının % 50-90’ında immunhistokimyasal olarak S-100 proteini eksprese edi[1]lir.
Fakat boyanma genellikle fokaldir. Diffüz S-100 po[1]zitifliği nadiren
konvansiyonel MPSKT’e ile bağdaşır ve selüler schwannom, melanom gibi diğer
tümörlerin ola[1]sılığını
arttırır. 1,10 MPSKT olgularının yarısını oluşturan NF1 hasta[1]ları,
nörofibromin isimli tümör baskılayıcı proteini kod layan NF1 geninin
(17q11.2) sadece bir kopyasına sa[1]hiptir.
Fonksiyonel olan bu kopyanın da somatik bir mu[1]tasyon sonucu etkisiz hale
gelmesiyle miyelinsiz Schwann hücre progenitörleri anormal büyüme göste[1]rerek,
benign nörofibromları oluştururlar. 3,4 Benign nö[1]rofibromlar sıklıkla
kozmetik sorunlara sebep olmasına rağmen pleksiform nörofibromlar ise yumuşak
dokuya ve kemiklere invaze olarak işlev bozukluklarına sebep olmaktadır.
Pleksiform nörofibromların yaklaşık %10’u, TP53 veya CDKN2A tümör baskılayıcı
genlerin kaybıyla beraber malign transformasyon sonucu MPNST’leri oluş[1]tururlar.
11 MPSKT’lerde hücrenin bölünmesi, büyümesi ve apoptozunu kontrol eden önemli
sinyal iletim yolakla[1]rındaki
bozulmalar sonucu çeşitli genetik anomaliler gö[1]rülür. Sinyal ileti
yolaklarındaki bu anomaliler TFF, EGFR, ARF, IGF1R gibi birçok önemli geni
içerir. 12 Bir[1]çok
MPSKT hastası çoklu sayısal ve yapısal kromozom anomalilerinden oluşan kompleks
karyotipe sahiptir. Karşılaştırmalı genomik hibridizasyon (CGH) analizleri
sonucuna göre kompleks karyotipler ortalama 18 abe[1]rasyon içermektedir, bu
aberasyonlarda genomik kazanç/ kayıp oranı 1:1’dir. Sıklıkla görülen
deoksiribonükleik asit (DNA) kopya sayısı değişiklikleri; 7p, 8q ve 17q ka[1]zancı
ile 9p, 11q, 13q ve 17p kayıplarıdır. 1 Literatürde kazanç ve kayıp
bölgelerindeki anlamlı genler bildiril[1]miştir.
Bu genler sırasıyla kazanç için; BIRC5, CCNE2, DAB2, DDX15, EGFR, MSH2, CDK6,
HGF, ITGB4, KCNK15, LAMA3, LOXL2, MET, PDGFRA genleri, kayıp için; CDH1,
GLTSCR2, EGR1, CTSB, GATA3, SULT2A1, LICAM2, MMP13, RASSF2 ve TP53 genleri[1]dir.

TEDAVİ
MPSKT
tedavisi diğer yumuşak doku sarkomlarında olduğu gibi cerrahidir. 15,16
Uygulanacak cerrahi, geniş ya da radikal rezeksiyon olarak yapılmalıdır. Bu nok[1]tada
amputasyon da bir seçenek olabilir. 6,17 Cerrahi işlem sırasında tümörün
kaynaklandığı sinir feda edi[1]leceği
için hastada morbidite oluşur. Bu yüzden gerekli bilgiler hastaya işlemden önce
verilmelidir. Paraspinal ve retroperiton yerleşimli büyük lezyonlu olgularda
geniş rezeksiyon yapılamayabilir ve lezyon subtotal olarak çıkarılmak zorunda
kalınabilir. Bu olgularda cerrahiye ilave olarak yüksek doz radyoterapi öneril[1]mektedir.
6 Kemoterapi ve radyoterapi de tedavinin diğer par[1]çaları arasında
sayılabilir. Radyoterapi ön plana çıkmakla beraber kemoterapinin etkinliği ve
yeri tartışmalıdır. 2 Volkan GÜRKAN ve ark. Turkiye Klinikleri J Orthop &
Traumatol-Special Topics 2017;10(4):325-30 327 RESİM 3: MPSKT’lerin
gösterilmesinde FDG PET oldukça duyarlıdır. Bu agresif tedavilerin lokal nüks
oranını azalttığı düşü[1]nülmesine
rağmen lokal nüks ve uzak organ metastaz[1]ları sıklıkla
gözlenmektedir. 6 PROGNOZ Prognozu belirleyen en önemli faktör; hastanın NF1
olup olmamasıdır. Bu grup hastalarda 5 yıllık sağ kalım oranı %53’den %16’ya
düşmektedir. Tümör çapının 5 cm’den büyük olması önemli bir diğer faktördür. Tümör
çapının büyük olduğu NF1’li hastalarda ise 5 yıllık sağ kalım %9’a
gerilemektedir. 16 Pozitif cerrahi sınır varlığı, lokal rekürens, yüksek
histolojik derece ve gövde yerle[1]şimi
diğer kötü prognostik göstergelerdir. 1,10 Metastazlar ise genellikle akciğer,
kemik, plevra ve retroperitona olur. Bölgesel lenf bezi tutulumu olguların
%9’unda göz[1]lenebilmektedir
DAMAR KÖKENLİ YUMUŞAK
DOKU SARKOMLARI
Damar
endotelinden kaynaklanan ve oldukça nadir gö[1]rülen bu tümörler;
hemanjiyoendotelyoma, Kaposi sar[1]komu
ve anjiyosarkom şeklinde sınıflandırılırlar. Bunların arasında anjiyosarkomlar
en kötü davranışlı olanı olup, mortalite oranı yüksektir. Diğerleri anjiyo[1]sarkomlar
kadar agresif seyir göstermez. Hemanjiyope[1]risitoma ise son dönemde
damar tümörleri sınıflamasın dan çıkartılarak ekstraplevral soliter fibröz
tümör adı al[1]tında
fibroblastik/miyofibroblastik tümörler sınıflama[1]sına dahil edilmiştir. 1,10
Hemanjiyoendotelyoma; hemanjiyom ile anjiyo[1]sarkom arasında kalan bir
lezyondur. WHO sınıflandır[1]masında;
epiteloid, kaposiform, retiform, kompozit ve papiller intralenfatik
anjiyoendotelyoma (Dabska tü[1]mörü)
gibi alt tiplere ayırılırlar. 1 Kaposi sarkomu; insan herpesvirüs 8 (HHV8) veya
Kaposi sarkom-ilişkili herpesvirüsün (KSHV) sebep ol[1]duğu kötü huylu bir cilt
lezyonudur. Ortaya çıkmasında viral nedenler ile beraber immünolojik, genetik
ve çev[1]resel
faktörler de rol oynar. Klasik/kronik, Afrika tipi en[1]demik, iatrojenik ve AIDS
ilişkili tipleri vardır. Klasik tipi Akdeniz ve Doğu Avrupa’da daha yaygındır.
Miyeloma, lösemi ve lenfoma gibi lenforetiküler neoplazmlar ile bir[1]likteliği
söz konusudur. Genellikle ileri yaşlarda gözlenir ve ölüm oranı %10-20
kadardır. İatrojenik tip, daha çok organ transplantasyonu sonrası ya da immün
baskılayıcı ilaç kullanımı ile ilişkilidir. Afrika endemik tipi, AIDS ile
ilişkili olmayıp, daha çok Ekvator bölgesinde yaşayan ço[1]cuklarda gözlenir. AIDS
bağlantılı tip ise en agresif formu olup daha çok yüz, genital bölge, alt
ekstremite, ağız içi mukozası ve lenf nodlarında gözlenir. 1,17
Anjiyosarkomlar; kötü huylu damar tümörleridir. Hemanjiyosarkom ve
lenfanjiyosarkomları kapsar. Bu gruplar arasında kesin ayırımın yapılması,
görüntüleme yöntemleri ve patoloji açısından çok zor olduğu için hep[1]sini
kapsayan anjiyosarkom terimi tercih edilmektedir. Yaşlılarda daha sık
görülürken, erkeklerde görülme sık[1]lığı
iki kat yüksektir. Oldukça nadir görülen bu tümör[1]ler tüm yumuşak doku
sarkomlarının ancak %1 kadarını oluştururlar. 17 Bununla beraber yumuşak doku
sarkom[1]ları
genellikle derin yerleşimliyken, anjiyosarkomlar daha çok yüzeysel yerleşim
gösterirler. En fazla deride (%33) görülmektedir. İç organlarda ve
kardiovasküler sistemde de görülebilirler. 2,5 Ayrıca lenfödemle ilişkili,
radyoterapi sonrası görülen ve meme anjiyosarkomları olarak bilinen tipleri
mevcuttur. Radyasyona sekonder olarak gelişen anjiyosarkomlar tüm vakaların
yaklaşık dörtte birini oluşturur. Pek çoğu yüksek doz radyasyon (ortalama 50
Gy) maruziyeti sonrası 5 yıl içerisinde ge[1]lişmektedir. 10
Anjiyosarkomlar, nadiren vücutta uzun süre kalan yabancı cisme (damar grefti ve
implant gibi) bağlı olarak da görülebilir. 2,7 Kronik lenfödem, anjiyo[1]sarkom
için bir yatkınlık oluştururken, vakaların sadece %10’unda böyle bir
birliktelik söz konusudur. 18 Konje[1]nital,
idiyopatik, travmatik ya da enfeksiyöz lenfödem[1]lerde de anjiyosarkom
gelişebilmektedir. 19 Ayrıca kronik osteomyelitte sinüs ağzında ya da kronik
bir yaranın et[1]rafında
da oluşabilir.
KLİNİK VE GÖRÜNTÜLEME
Kötü
huylu damar tümörleri derin yerleşimli olduğu zaman farklı görüntüler sunar.
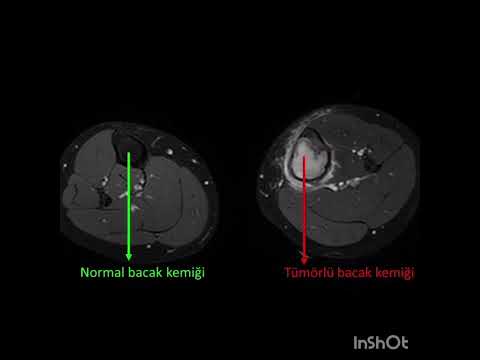
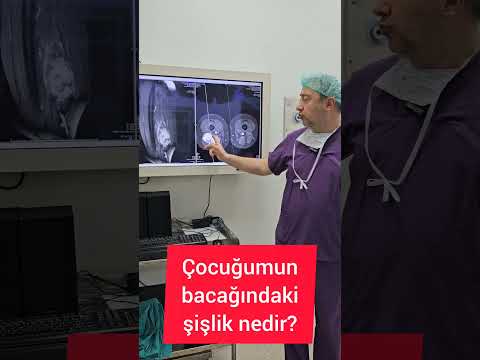
Lezyon kemiğe yakın yerleşmişse direkt grafilerde, BT ve MR’da kemik des[1]trüksiyonu
gözlenebilir (Resim 5). Ayrıca kalsifikasyon da dikkat çekebilir. Bu lezyonları
birbirinden ayırt etmek zordur. Çoğu zaman büyük bir damarla ilişkili in[1]termusküler
kitleler şeklinde kendini gösterir. Bu lez[1]yonların ultrasonografi
(USG), bilgisayarlı tomografi (BT) ve MR’da spesifik bulguları yoktur. USG’de
hipo/hiper ekoik olabilir ve kistik alanları bulunabilir. MR, görüntü[1]lemede
en üstün yöntemdir. MR ile özellikle kitlenin pe[1]riferinde yer alan damar
kanalları çok iyi bir şekilde görüntülenebilir. Bu kanallarda çoğu zaman yüksek
akım bulunur. Diğer yumuşak doku sarkomlarında –ki bunlar hipervasküler bile
olsalar, bu şekilde damar benzeri ka[1]nallar
yer almaz. 5 Kötü huylu damartümörlerinin MR gö[1]rüntülemesinde T1’de orta
dereceli birintensite, T2’de ise hiperintensite söz konusudur. Hemanjiyomlar
vasküler

kanal ve boşluklar içerdiği için gün
içerisinde çapları de[1]ğişiklik
gösterebilir. Ancak kötü huylu damar tümörleri[1]nin solid komponentleri
daha ön planda olduğu için bu bulgu görülmemektedir (Resim 4 ve 5). Anjiyografi
ise bu lezyonların gösterilmesinde yardımcıdır. Hemanjiomlarda çoğu zaman
klinik özellikler ve tipik MR ve USG bulgularıyla tanıya gitmek mümkün olsa da
damar kökenli habis yumuşak doku tümörleri için aynı durum söz konusu değildir;
tanı, mutlaka bi[1]yopsi
ile konur. Hemanjiomlara göre daha az kistik ya[1]pıda olduklarından iğne
biyopsisi sonucu aşırı kanama olasılıkları daha düşüktür. Aşırı kanama olsa
bile biyopsi yerine bir süre basınç uygulanarak bu durumun önüne geçilebilir.
Ancak özellikle derin yerleşimli anjiyomatöz lezyonlarda iğne biyopsisinden
sonra aşırı kanamaya bağlı ölüm nadiren de olsa bildirilmiştir. 5 Anjiosarkom[1]ların
patolojik tanısının konması zordur. Bu durum bir[1]kaç kez biyopsi tekrarını
gerektirebilir. PATOLOJİ VE GENETİK Anjiyosarkomlar makroskopik olarak sıklıkla
sekonder kistik dejenerasyon ve nekroz içeren, multinodüler, he[1]morajik
kitlelerdir. Yumuşak doku anjiyosarkomlarının morfolojisi iyi yapılanmış,
anostomozlaşan damarlardan, yüksek dereceli epitelioid veya iğsi hücrelerden
oluşan solid tabakalara kadar değişebilir. Çoğunlukla yüksek mitotik
aktivitesi, koagülatif nekrozu ve belirgin nük[1]leer atipisi ile yüksek
dereceli tümörlerdir. 1,9 Epitelioid morfolojili anjiyosarkomlar, karsinom veya
melanom ile ayırıcı tanı güçlüğü oluşturabilir. 9 İmmunhistokimyasal olarak
tipik vasküler belirteçler olan CD34, CD31, ERG ve FLI-1 ile boyanırlar. 1,9
Anjiyosarkomlar hakkındaki genetik çalışmalar ye[1]tersizdir ve vaka sayısı da
sınırlıdır. Bu tümörler genel[1]likle
kompleks karyotip göstermektedir. Henüz net olarak tanımlanmış ya da süreklilik
gösteren bir kromo[1]zom
anomalisi ortaya konamamıştır. Sitogenetik olarak incelendiğinde farklı
bölgelerde görülen anjiyosarkom[1]larda
benzer kromozomal özellikler tespit edilmiştir. En sık rastlanan değişiklikler;
5pter-p11, 8p12-qter, 20pter[1]q12
bölgelerinde kazanç ve 4p, 7p15-pter, 7p15-Y böl[1]gelerinde kayıp ve spesifik
olarak 22q bölgesini içeren anomalilerdir. Akım sitometri kullanılarak
gerçekleşti[1]rilen
genetik çalışmalarında diploid, tetraploid ve anöp[1]loid hücre paternleri
gösterilmiştir. Ancak bu bulguların klinikle ilişkisi gösterilememiştir. 14,20
Anjiyosarkomlar için gerçekleştirilen genetik çalışmalara göre TIE1, KDR, TEK
ve FLT1 genlerini içeren spesifik vasküler tirozin kinaz reseptörlerinin diğer
sarkom tiplerine oranla eks[1]presyonlarının
arttığı gösterilmiştir.
TEDAVİ
Orta
dereceli agresif olan hemanjiyoperisitoma ya da he[1]manjiyoendotelyomanın
tedavisinde genellikle geniş re[1]Volkan
GÜRKAN ve ark. Turkiye Klinikleri J Orthop & Traumatol-Special Topics
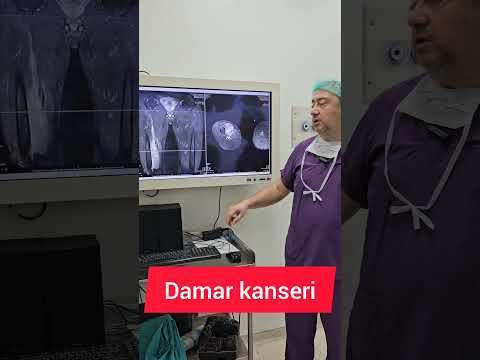
2017;10(4):325-30 329 RESİM 4: Uyluk yerleşimli bir anjiyosarkom olgusunda T2
sekans koronal plan MR görüntüsü. Prof.Dr. Harzem Özger’in arşivinden,
kendisinin özel izni ile alınmıştır. RESİM 5: Aynı hastanın aksiyal MR
kesitinde lezyonun solid komponenti dikkat çe[1]kiyor. Ayrıca lezyonun femurda
destrüksiyona neden olduğu görülüyor. Prof.Dr. Harzem Özger’in arşivinden,
kendisinin özel izni ile alınmıştır. zeksiyon yeterlidir. Ancak bunlarda lokal
nüks görüle[1]bilir
ve olguların %10-25’inde en fazla akciğer, kemik ve karaciğere olmak üzere uzak
organ metastazları gözle[1]nebilir.
Anjiyosarkomun tedavisi çok daha agresif olma[1]lıdır. Burada agresif
geniş/radikal rezeksiyon ve adjuvan radyo/kemoterapi uygulanır. Buna rağmen
prognoz çok daha kötüdür. Bazen ameliyattan hemen sonra operas[1]yon
yerinden sürekli bir kırmızı-kahverengimsi kanama olur ve bu durum uzun
sürebilir. Lokal nüks sıktır ve diğer yumuşak doku sarkomlarından daha kısa bir
süre içerisinde meydana gelir. Anjiosarkomlar büyük ölçüde kemoterapiye
dirençli tümörlerdir. Kemoterapinin sağ kalım üzerine bir etkisi
gösterilememiştir. 19 Ancak me[1]tastatik
hastalarda kemoterapi ile hastalığın ilerleme[1]sinde yavaşlama sağlandığı
düşünülmektedir. Antianjiyogenetik ajanlar bu noktada etkin olabilmekle beraber
daha fazla çalışmaya ihtiyaç vardır. 19,22 PROGNOZ Kaposi sarkomunda prognoz,
lezyonun tipi ile ilişkilidir. AIDS bağlantılı tipi en kötü prognoza sahiptir.
Anjiyo[1]sarkomlarda
ise kötü prognostik faktörler; ileri yaş, rad[1]yasyona sekonder gelişen
olgular, retroperitoneal yer leşim, tümör boyutunun büyük olması, yüksek Ki67
oranı ve pozitif cerrahi sınır varlığıdır. 1,10 Akciğer, kemik ve lenf
nodlarına erken metastaz görülebilir. Lenfödemle beraber olan anjiyosarkomlarda
5 yıllık sağ kalım %15’dir. Derin yerleşimli lezyonda ise hastaların %53’ü bir
sene içerisinde kaybedilir.